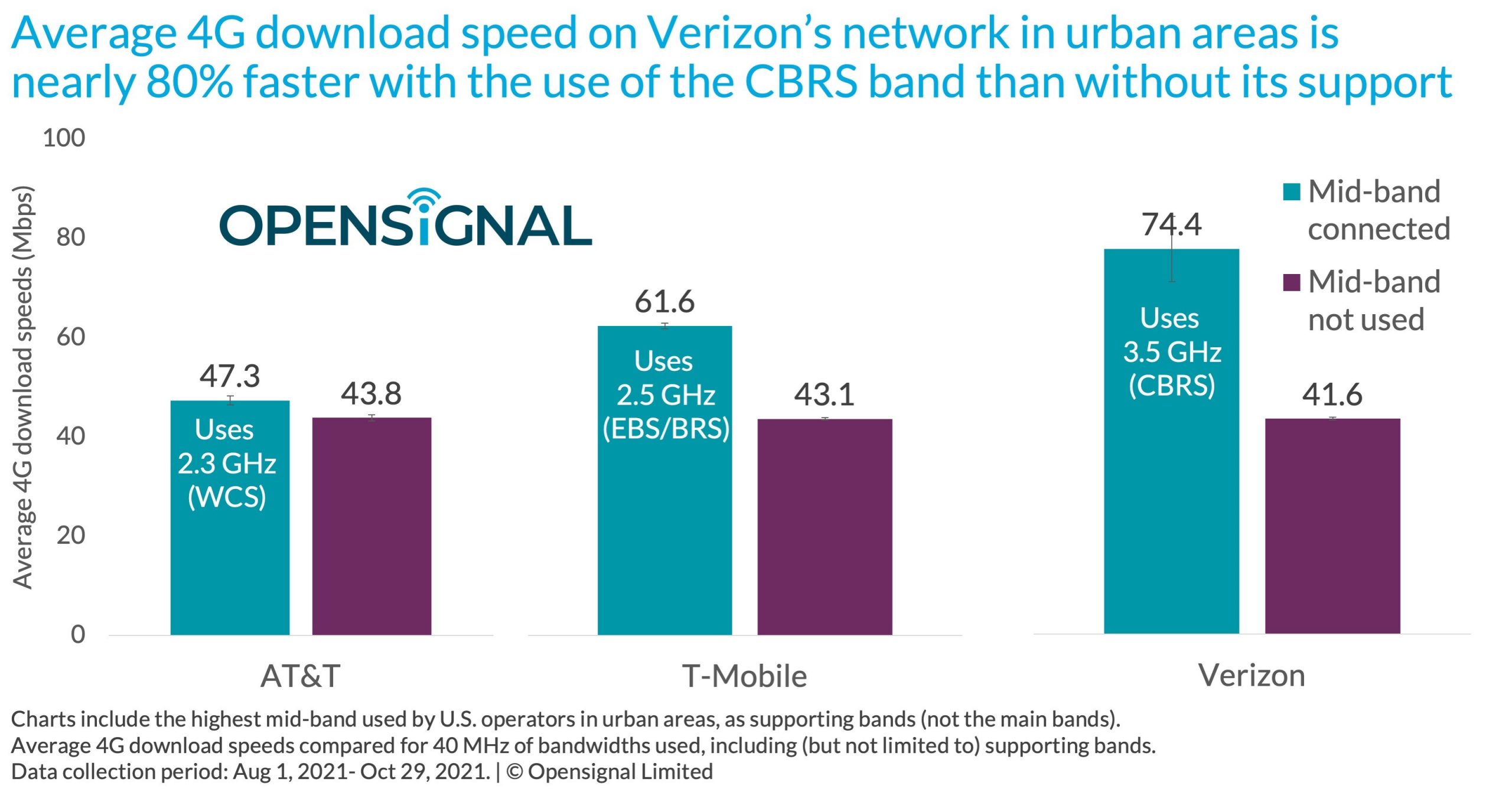
约翰斯 · 霍普金斯大学一支团队开发的 UNCALLED 软件纳米孔测序取得突破,解码基因组所需要的时间大大减少,由过去的 15 天缩短至 3 天甚至更少。
还记得电影《黑客帝国》中,当 Neo 释放他的全部力量时,周围的世界以各个方向运行的代码行显示的场景吗?如果你能够以这种方式观察周围的世界,则坐在你旁边的人显示为一个网页,你可以单击右键以检查元素和找到隐藏的源代码。
 图源:https://www.youtube.com/watch?v=Vy7RaQUmOzE
图源:https://www.youtube.com/watch?v=Vy7RaQUmOzE虽然电影中的场景在现实中依然实现不了,但借助于开源软件,纳米孔测序研究在最近实现了突破,解码基因组所需要的时间大大减少,由过去的 15 天缩短至 3 天甚至更少。不久之前,解码一个基因组需要数年时间。这一切都要归功于此前约翰斯 · 霍普金斯大学一支团队开发的 UNCALLED 软件。
项目地址:https://github.com/skovaka/UNCALLED
为了了解 UNCALLED 背后的代码,Stack Overflow 对约翰斯 · 霍普金斯大学计算机科学与生物学 Bloomberg 杰出副教授、DNA 测序数据大规模计算检测专家 Michael Schatz 进行了深入访谈。
 图源:https://hub.jhu.edu/2016/05/26/michael-schatz-bloomberg-distinguished-professor/
图源:https://hub.jhu.edu/2016/05/26/michael-schatz-bloomberg-distinguished-professor/首先来看纳米孔测序仪。Schatz 表示,「这个想法起源于 30 年前,据说是在餐巾纸上绘制的第一个图表。」实际上,纳米孔测序的概念最初是由 David Deamer 博士使用红色圆珠笔勾勒出来的。

想象一下,一条单链 DNA 可以一次穿过一个很小的孔。将遗传物质穿过这个小孔,并且组成人类基因组的 As、Ts、Gs 和 Cs 将依次显示出来。问题来了,如何区分 DNA 的四个组成部分呢?
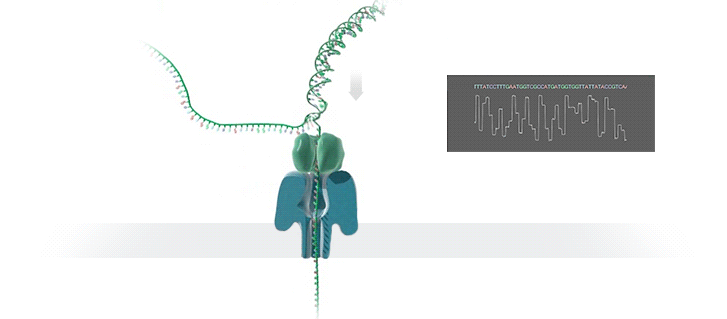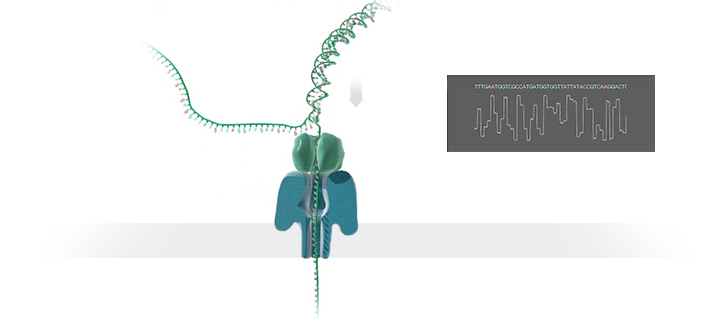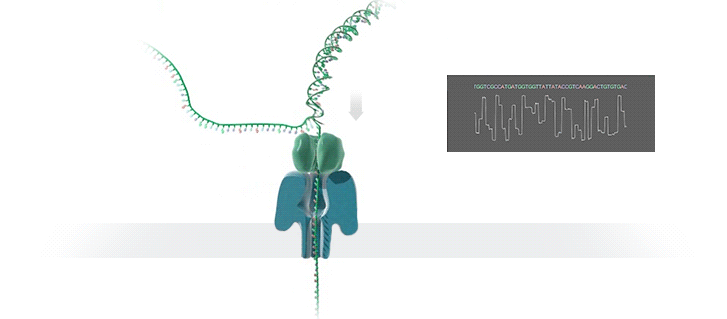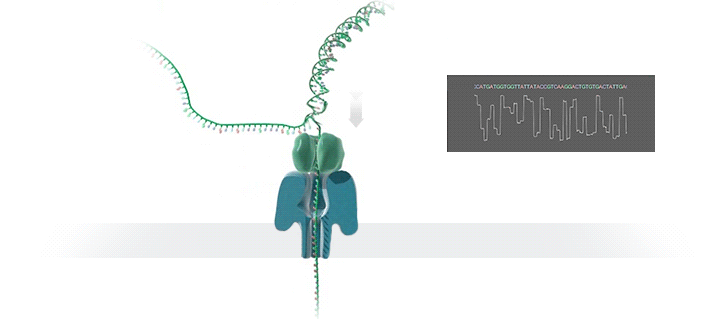 图源:Oxford Nanopore
图源:Oxford NanoporeSchatz 解释道,「这就需要你能想象到的最精细的测量,测量与不同 bits DNA 相关的电流变化。这需要微微安培级别的测量——安培的万亿分之一。现在,我们可以实时获得这些读数了。」五年前,这类工作需要的设备仅能够在正式的研究机构中配备。但是现在,你只需要花 1000 美元,就可以买到一个纳米孔测序仪作为外围设备,并通过 USB 连接到任何计算机上。
测序还会产生非常嘈杂的电子数据,对此 Schatz 及其团队开发了一种受马尔可夫模型启发的模糊逻辑(fuzzy logic),以接近实时的方式解码每个蛋白质。Schatz 兴奋地说道,「当核苷酸通过这个小孔时,我们可以每秒对电流进行 4000 次测量。」
测序会产生非常嘈杂的电子数据,但 Schatz 和他的团队开发了一种受马尔可夫模型启发的模糊逻辑,可以近乎实时地解码每种蛋白质。Schatz 表示「 我的意思是,这基本上是出自《星际迷航》,核苷酸通过这个小孔,我们每秒测量电流四千次。」该软件正在实时解码序列,以便将其与不同的遗传标记相匹配。
通过这个小孔的每一位 DNA 都是一个带电分子。该软件允许用户实际反转单个分子上的电压,从而将其从纳米孔中弹出。正是这种选择性地仅对与手头工作相关的部分进行排序的能力,才能够大幅提高速度。
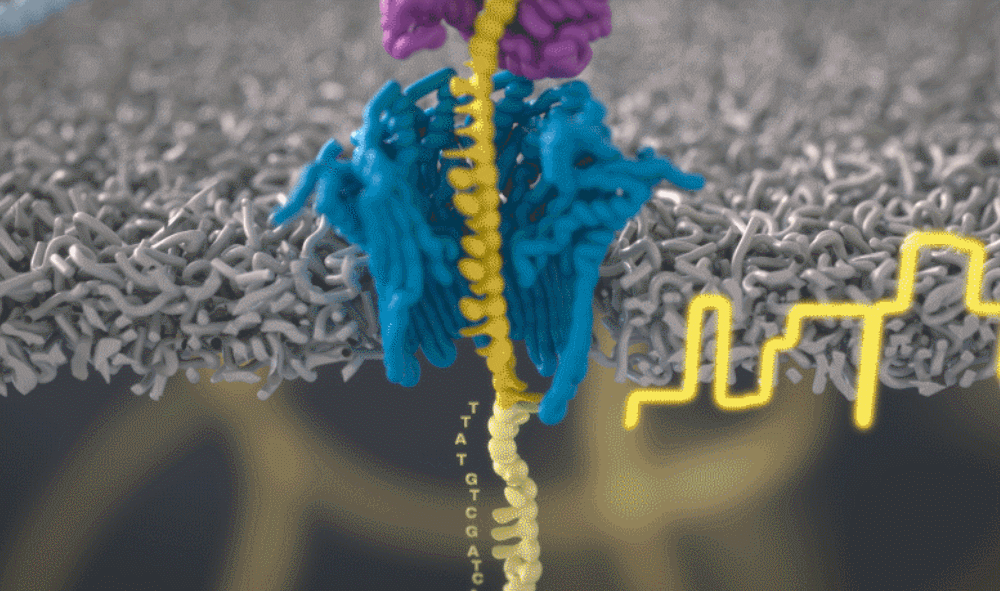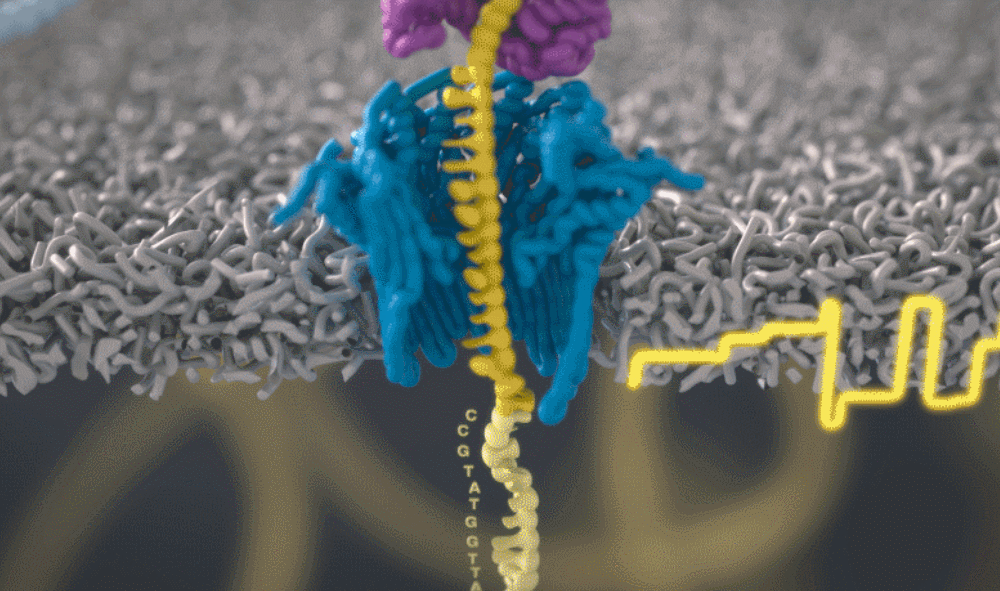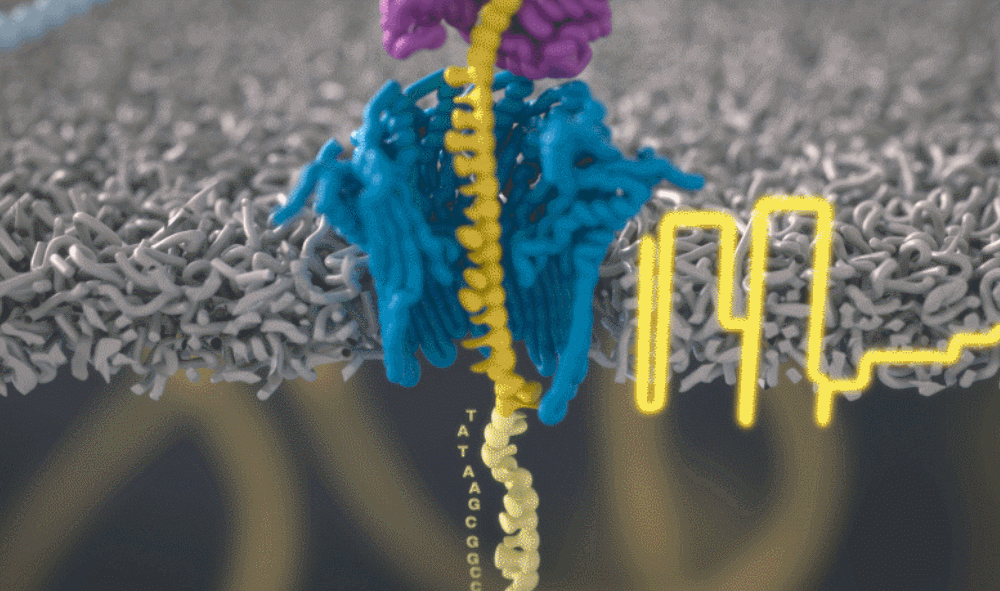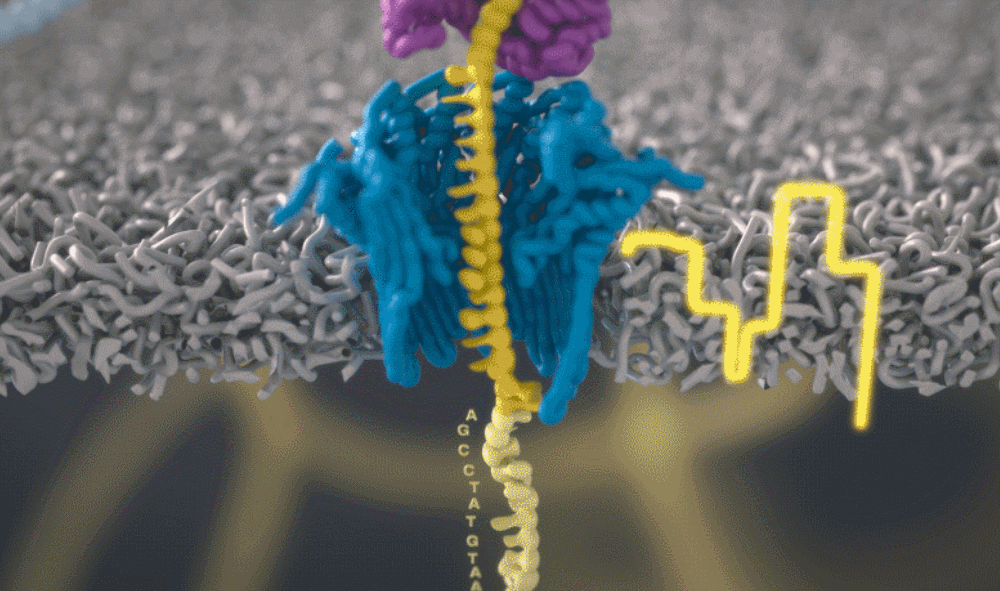 图源:Oxford Nanopore
图源:Oxford NanoporeUNCALLED 背后的代码
每个 DNA 片段都会根据核苷酸返回电压读数。对于电流数据,你可能需要的是 A 核苷酸特定的电流数据,而需要 C 核苷酸不同的电流等,但是,我们怎么读懂这些数据?
电流实际上与连续的几个核苷酸有关,大约有六个核苷酸对电流影响最大。所以实际上你在不同的环境中,在 6 个核苷酸周围,大约 6 次感觉到相同的核苷酸。对于特定电流测量,通常会涉及数百个核苷酸序列。
想想这六个组合的每个组合都有一个偏移量。在第一偏移量处,有一百个可能的核苷酸序列;在偏移二处,同样也有一百个可能的核苷酸序列;以此类推,在其他偏移量处,存在一百个可能的核苷酸序列。但正是在重叠序列的组合中,我们才有希望将其解析为特定的核苷酸,因为我们知道序列必须重叠。例如,偏移量 1 处的 GATTACA 之后可以是偏移量 2 处的 ATTACAT,但不能是 TTTACAT、AATACAT 或任何其他不以 ATTACA 开头的序列解码。
获得核苷酸序列后,你需要进行文本处理以确定该分子源自基因组中的哪个位置。目前有一种非常强大的数据结构可以使用,称为 Burrows–Wheeler transform,如今它已成为基因组学的核心。该算法于 1994 年被 Michael Burrows 和 David Wheeler 在位于加利福尼亚州帕洛阿尔托的 DEC 系统研究中心发明。它的基础是之前 Wheeler 在 1983 年发明的一种没有公开的转换方法。
相比于几年前的实验室工具,纳米孔测序仪非常便宜。该软件的作用是,我们不必扫描整个基因组,而是针对特定分子进行测序,Schatz 表示:我们可以实时选择哪些分子会被完全读出,哪些分子会在大约一秒钟的测序后读出。
因此,假如你想确定一个人是否携带与遗传性癌症相关的基因中的变异,例如 BRCA1,你需要采集样本。如果你想用纳米孔测序来分析所有材料,那将是一个非常缓慢和昂贵的过程。所有的分子都在试管中混合在一起,当随机从该集合中取出分子时,你可以对它们进行排序。然而,Schatz lab 研发的新软件 UNCALLED,可以近乎实时地评估一个序列是否值得研究,该软件由博士生 Sam Kovaka 领导完成。
事实上,在正常序列中,你可能希望对基因组进行多次测序,因为你采集的任何样本都有随机的 DNA 分子集合,并且可能不包含最感兴趣的部分。假如可以进行选择,你可以更快地筛选你想要的东西,避免一次又一次地排序其他领域。
我们以传染病为例, 随着测试的激增,世界各地的实验室都在努力应对大量工作负载。在那种情况下,人类基因组可能贡献不大,因为那不是你真正要找的。有了 UNCALLED,纳米孔就会喷射出关于人类相关的东西。任何与人类基因组不匹配的东西,我们都会舍弃,这样就可以对其进行一些实时分析。
当 Schatz 首次进入基因组学领域时,该行业存在的最大问题是封闭和技术私有。在很早的时候,研究者努力进行大量基因专利申请。例如,与乳腺癌相关的基因研究案例。研究者为这些序列申请专利,并收取大量使用资金。
幸运的是,这一趋势近年来有所好转。
Schatz 表示:能够共享代码和协同工作有很多好处。在不同的场景下,开源带来的好处超过了其所带来的负面影响。
英文原文:https://stackoverflow.blog/2021/12/24/sequencing-your-dna-with-a-usb-dongle-and-open-source-code/