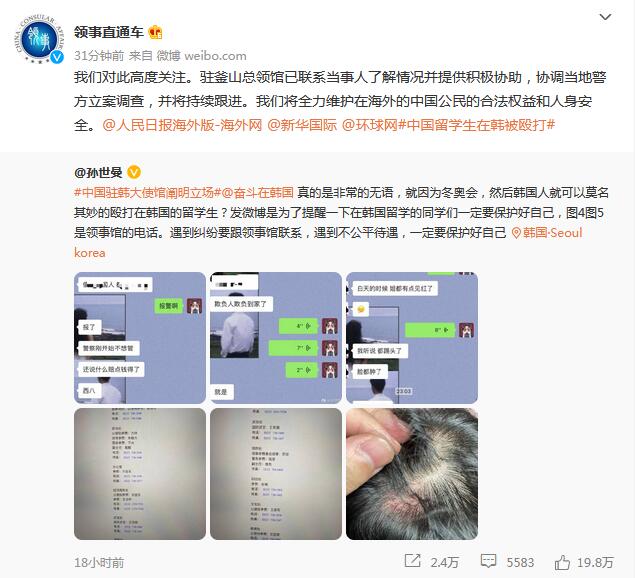
美国食品药品监督管理局FDA委员会周四投票建议不要完全批准信达生物和礼来(Eli Lilly)开发的肺癌治疗药物信迪利单抗(sintilimab),理由是担心该临床试验仅在中国进行,参与者不像美国人口那样多样化。
FDA肿瘤药物咨询委员会(ODAC)以14比1的投票结果,要求企业应在获得最终批准之前进行更多反映美国患者的临床试验。信达生物(1801.HK)周五收盘股价大跌超过6%。
FDA肿瘤学卓越中心主任帕兹杜尔(Richard Pazdur)博士澄清,这一结果目标不是排斥中国,而是强调将药物开发纳入国际范围的重要性。
但除了临床数据需要更多样化之外,FDA还强调了企业在细节披露方面的疏忽。ODAC认为,该试验未能达到FDA的知情同意标准,因为它没有明确列出经批准的疗法或参与替代研究的治疗方法。
“虽然数据完整性在临床研究中至关重要,但伦理方面的完整性更为重要。”美国国家癌症研究所临床主任马丹(Ravi Madan)教授表示。FDA认为,企业的知情同意书在试验期间没有根据要求更新。
一位曾在美国FDA工作的中国专家告诉第一财经记者:“之所以FDA要求递交知情同意书,是为了给病人入组完整的信息,让病人知道当时的治疗方法的情况背景,这样患者可以判断是否接受入组研究。”
信达生物和礼来也在回应中表示:“申办方应该更新知情同意书,这样(临床试验)中心才有机会根据各自的政策和流程,来更新中心层面的知情同意书。”
企业在知情同意书方面的疏漏也引发大量的评论,并提醒中国企业将来在做临床研究时,一定要更加注重完善细节。
此外,企业与监管机构在临床研究的前期沟通也很重要。FDA方面称,该研究自2018年8月启动以来,整个临床试验过程中,企业几乎没有与FDA进行过任何沟通咨询。一直到2020年4月,该药物要申请上市批准时,FDA才收到通知,当时两家公司提交了临床试验的结果并计划寻求申请批准。
一位与FDA长期合作的药企方面人士对第一财经记者表示:“申请人和申办者在试验设计早期,与监管机构缺乏接触沟通令人感到失望,我相信如果这些必要的接触能够尽早进行,而不是等到药物要批准的最后一刻才进行,那么结局可能会不一样。”
上述人士认为,与监管机构建立起畅通的对话非常重要,要让监管方对临床试验进展保持知情,这有助于提高药物获批成功的几率。
“与FDA多年交往,我认为他们的专家是公正的并且严谨的,而且效率很高。”他补充道,“企业在策划临床设计方案时候就需要提前与FDA沟通,这种沟通没有成本,而且专家都很耐心,可以帮助企业少走弯路。”
但无论如何,这项申请还是为中国药物向海外进军迈出了重要一步,也给未来希望在海外上市新药的中国企业提供了宝贵的经验和教训。
“中国生物医药行业目前的发展已经与几年前无法同日而语。”生物基金公司Loncar Investments创始人布拉德•隆卡(Brad Loncar)对第一财经记者表示,“信达和礼来虽然闯关失败,错失了与美国的K药(Keytruda)和O药(Opdivo)在全球市场竞争的机会,但中国生物制药企业近年来研发能力有了很大提升,未来一定会引领全球。”