今天,分享一篇哈佛团队开发罕见变异荟萃分析方法,为整合分析海量基因组数据提供有效方案,希望以下哈佛团队开发罕见变异荟萃分析方法,为整合分析海量基因组数据提供有效方案的内容对您有用。
来源:DeepTech深科技
厉希豪,目前是哈佛大学陈曾熙公共卫生学院生物统计系研究员,师从美国国家医学院院士、哈佛大学生物统计系林希虹教授。
 图丨厉希豪(来源厉希豪)
图丨厉希豪(来源厉希豪)据介绍,由于全基因组测序成本的逐步降低,包括美国国家心肺血液研究所 20 万人的精准化医学研究计划(Trans-Omics Precision Medicine Program, TOPMed)、50 万人的英国生物样本库(UK Biobank)等在内的大规模全基因组测序研究,正在迅速开展。
将这些经由测序得到的高通量基因组学数据,与其他生物医学大数据相结合,可以有效地定位致病基因与遗传位点、预测个体或群体对特定疾病的遗传风险,进而在揭示各种复杂疾病的分子机制、开发新的药物标靶,以及制定精准的个性化健康诊疗策略等方面发挥重要作用。
与此同时,由于遗传信息具有高度敏感性,个体的全基因组测序数据是受到高度保护并被限制共享的。比如,UK Biobank 就要求所有研究人员,在其指定授权的研究分析云计算平台上,进行全基因组测序数据的处理与分析。
因此,如何有效地整合并集中分析这些数据,并使各自研究中的个体基因组数据得到保护,就成为了一个亟待解决的问题。
对此,该团队将荟萃分析这种统计学方法,用于全基因组测序研究中,开发了大规模全基因组测序数据罕见变异荟萃分析方法 MetaSTAAR,实现了全基因组汇总统计数据的高效存储共享和功能知情的荟萃分析,为整合这些大型测序研究中的基因组数据并保护个体数据隐私,提供了有效的解决方案。
具体而言,该方法的使用流程主要分为两大步。
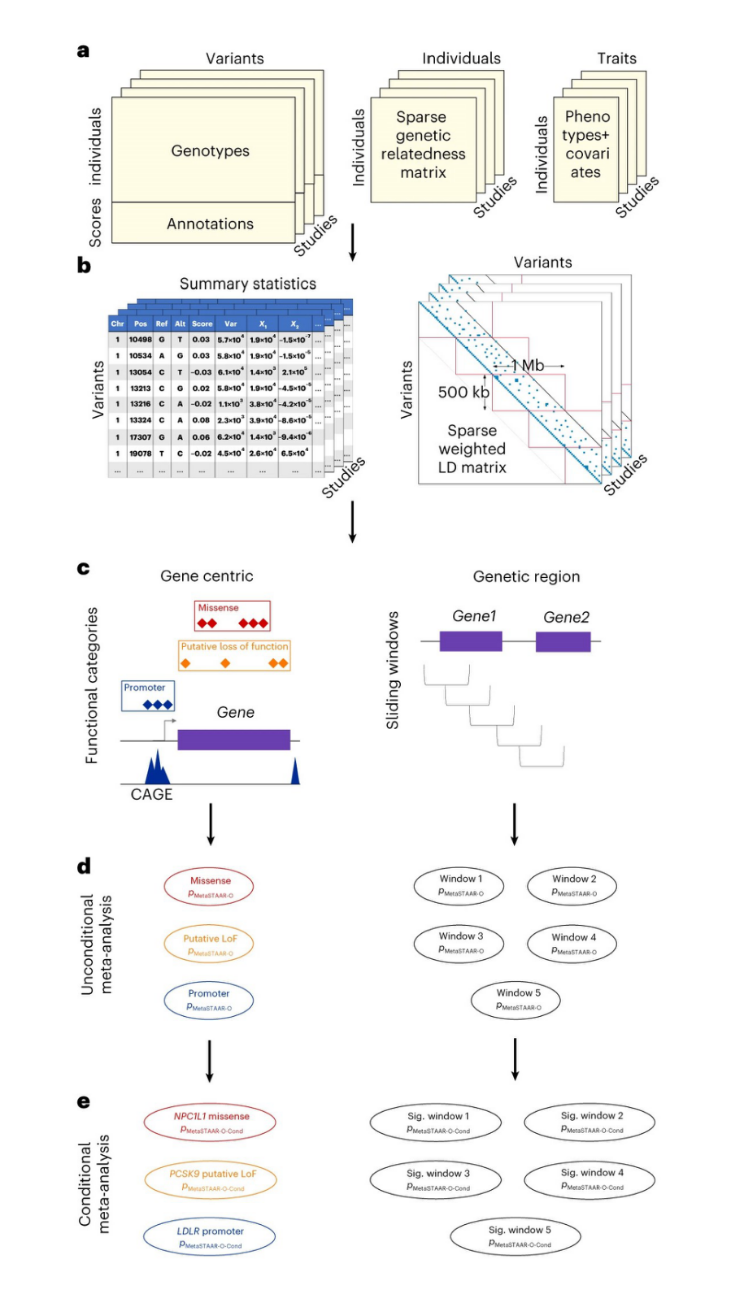 图丨 MetaSTAAR 荟萃分析流程(来源:Nature Genetics)
图丨 MetaSTAAR 荟萃分析流程(来源:Nature Genetics)第一步,参与荟萃分析的每项研究,通过使用 MetaSTAAR 的子工具 MetaSTAARWorker,对其全基因组测序数据生成汇总统计数据,包括每个遗传变异的得分统计量信息和遗传变异之间的协方差矩阵信息,同时存储并在不同的研究之间共享。
第二步,使用 MetaSTAAR 工具整合参与研究的汇总统计数据,并进行功能知情的荟萃分析。
“需要特别指出的是,在第一步中,参与荟萃分析的每项研究所共享的汇总统计数据是可以保护个体数据隐私的,这也是 MetaSTAAR 作为一种罕见变异荟萃分析方法的意义所在。”厉希豪说。
那么,与现有用于遗传关联研究中罕见变异荟萃分析的方法相比,MetaSTAAR 又具备哪些方面的优势呢?
他表示:“我们这项研究为全基因组测序数据荟萃分析提供了高效和高性能的分析方法,突破了大规模全基因组测序研究汇总统计数据存储瓶颈和荟萃分析运算瓶颈,填补了大规模全基因组测序数据中罕见变异荟萃分析方法的空白。”
据介绍,其和团队在发展 MetaSTAAR 时,面临了来自方法研究、数据分析等多方面的挑战。
比如,其在分析 TOPMed 约 30000 人的全基因组测序数据中,发现遗传变异的数量高达约 2.5 亿个。这就要求所发展的统计方法和具体的算法,能够在现有的计算能力下完成对海量数据的处理,并生成大小合理的汇总统计量,方便不同研究之间共享。
“在林希虹教授的指导和李子林博士的帮助下,我们在 MetaSTAAR 的研发早期,对方法和工具进行了大量的迭代,最终实现了比同类方法节省了数百倍的存储空间。”厉希豪说。
后面,他和团队希望基于 MetaSTAAR 做进一步的创新,不仅计划发展适用于不同表型的荟萃分析方法,比如多表型联合分析,生存数据表型分析等,还计划将高通量全基因组测序数据与其他生物医学大数据做进一步整合,发展出一套高效、开源、易使用、可扩展的方法。
课题组期待在为世界各地的科研人员提供必要且有效的分析方法与工具的同时,也为生物统计学、统计遗传学和基因组学的发展,做出自己的一份贡献。
参考资料:
1. Li, X., Quick, C., Zhou, H. et al. Powerful, scalable and resource-efficient meta-analysis of rare variant associations in large whole genome sequencing studies. Nature Genetics 55, 154–164 (2023). https://doi.org/10.1038/s41588-022-01225-6
2. Li, X., Li, Z., Zhou, H. et al. Dynamic incorporation of multiple in silico functional annotations empowers rare variant association analysis of large whole-genome sequencing studies at scale. Nature Genetics 52, 969–983 (2020). https://doi.org/10.1038/s41588-020-0676-4